Purpura thrombopénique immun (PTI)
Brève description:
Dans cette affection, la thrombopénie est due à des auto-anticorps anti-plaquettaires. On distingue une forme aiguë et une forme chronique.
Aspect clinique:
Le purpura thrombopénique immun aigu se rencontre surtout chez l'enfant suite à une infection virale aspécifique. Les hémorragies sévères sont exceptionnelles. L'évolution est spontanément favorable en quelques semaines dans 90% des cas. La forme chronique est plus fréquente chez l'adulte, en particulier chez les jeunes femmes (sex ratio de 1:3). Généralement, la maladie se développe à bas bruit. Les hémorragies sévères sont rares, même lorsque les valeurs plaquettaires sont inférieures à 10 x 109/l. Les principales manifestations cliniques sont des pétéchies généralisées et des hématomes spontanés, c'est à dire sans notion de traumatisme déclenchant (easy bruising: bruise, en anglais = bleus). Le traitement initial repose sur l'administration de corticostéroïdes; en cas d'échec, la splénectomie peut s'avérer efficace.
Le purpura thrombopénique immun est idiopathique ou secondaire (syndromes lymphoprolifératifs, collagénoses).
Aspect hématologique:

Le sang périphérique est caractérisé par une thrombopénie et une anisocytose plaquettaire.
Aspect de la moelle osseuse:
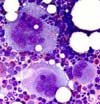
On trouve classiquement une augmentation des mégacaryocytes.
Thérapie:
Le traitement consiste d’abord en corticostéroïdes per os ou intraveineux. En cas d’urgences hémorragiques, des immunoglobulines intraveineuses peuvent entraîner une augmentation rapide de la numération thrombocytaire. La réponse thérapeutique aux immunoglobulines ne se maintient cependant que quelques semaines. Dans les cas réfractaires ou les récidives, la splénectomie ou une immunothérapie avec l’anticorps anti-CD20 rituximab (Mabthera®) sont les options thérapeutiques disponibles. Les formes chroniques peuvent être traitées par des agonistes du récepteur de la thrombopoïétine, tels que l’eltrombopag (Revolade®) ou le romiplostim (Nplate®).