Thrombocytémie essentielle (TE)
Brève description:
La thrombocytémie essentielle fait partie des syndromes myéloprolifératifs. Elle est caractérisée par des valeurs plaquettaires supérieures à 600 x 109/L de manière persistante. Des comptes 15 fois plus élevés que la limite supérieure des intervalles de référence sont possibles. Les fonctions plaquettaires sont fréquemment perturbées avec pour conséquence des complications hémorragiques ou thrombotiques.
Aspect clinique:
La thrombocytémie essentielle touche des individus dans la soixantaine. Elle est souvent asymptomatique pour une longue durée, si bien que le diagnostic est parfois évoqué fortuitement. La symptomatologie, lorsqu'elle apparaît, est en rapport avec des anomalies de la microcirculation: troubles neurologiques fluctuants tels que érythromélalgie, céphalées et vertiges. Une acrocyanose et des lésions ischémiques (par exemple, accidents vasculaires cérébraux) sont imputables à des thromboses artériolaires.
En raison des risques hémorragiques et thrombotiques, les options thérapeutiques ne sont pas toujours évidentes. Lors de numérations plaquettaires supérieures à 1'000 x 109/L, une cytoréduction s'impose; elle fait appel à l'Hydroxyurée, à l'Anagrelide ou à l'interféron α. En présence d'accidents thrombotiques, on prescrira des antiagrégants plaquettaires (par exemple, Aspirine), en étant conscient qu'une complication hémorragique n'est pas exclue.
Mécanisme pathogénique:
La mutation JAK2 V617F, MPL et les mutations CALR sont des mutations dites «driver», qui induisent directement le phénotype d’une mégacaryopoïèse augmentée. Les récepteurs de la thrombopoïétine MPL et JAK2 font partie de la principale voie de signalisation activatrice de la mégacaryopoïèse. Le mécanisme précis des mutations CALR n’est pas encore connu. On admet aujourd’hui qu’il faut chez au moins une partie des patients d’autres mutations pour déclencher la maladie. Ce qui n’est pas encore bien compris, c’est pour quelle raison des mutations identiques peuvent donner lieu à des maladies différentes (MFP, TE ou PV).
Traitement:
Pour diminuer le risque de complications thromboemboliques, il faut réduire le nombre de thrombocytes. On recourt pour cela à l’hydroxyurée (Litalir®), à l’anagrélide (Xagrid®, Thromboreductin®) ou à l’interféron alpha.
Aspect hématologique:
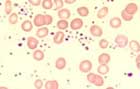
Le nombre de plaquettes est augmenté (valeurs > 600 x 109/l). On observe morphologiquement une anisocytose plaquettaire, la présence de plaquettes géantes et des fragments nucléaires de mégacaryocytes. Une augmentation des basophiles est fréquente. La thrombocytémie essentielle doit être distinguée des thrombocytoses réactionnelles (par exemple, lors d'anémie par carence en fer ou de cancers) et des thrombocytoses entrant dans le cadre d'autres syndromes myéloprolifératifs comme notamment la leucémie myéloïde chronique (recherche du chromosome Philadelphie ou du transcrit de fusion bcr-abl).
Aspect de la moelle osseuse:
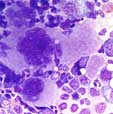
La mégacaryopoïèse est légèrement à fortement augmentée avec de grandes formes fortement hyperlobulées (noyaux mégacaryocytaires en «cornes de cerf» = staghorn like). On trouve à la biopsie des mégacaryocytes en groupes relativement relâchés, mais pas en amas serrés. Les cellules sont plutôt monomorphes et les formes de petite taille ne sont pas présentes en nombre. La cellularité de la moelle est typiquement normale, mais peut aussi être légèrement augmentée en cas de TE (thrombocytémie essentielle) JAK2 V617F positive. Une cellularité fortement augmentée parle contre le diagnostic de TE et est plutôt compatible avec une phase hypercellulaire d’une myélofibrose primitive. Une fibrose de réticuline ou de collagène significative parle aussi contre un diagnostic de TE.
Diagnostic:
D’après la classification OMS 2016, le diagnostic de thrombocytémie essentielle peut être retenu lorsque les quatre critères majeurs ou les trois premiers critères majeurs et le critère mineur sont vérifiés:
Critères majeurs:
- Thrombocytes >450 x 109/l
- La prolifération de la lignée mégacaryocytaire prédomine dans la biopsie de moelle avec une multiplication de mégacaryocytes matures à noyaux hyperlobulés. Pas d’augmentation significative ni de déviation gauche de la granulopoïèse ou de l’érythropoïèse avec très rarement présence de discrets foyers de fibrose (grade 1)
- Non vérification des critères OMS pour LCM BCR-ABL11, PV, MFP, syndromes myélodysplasiques ou autres néoplasies myéloïdes
- Mise en évidence de mutation JAK2, CALR ou MPL
Critères mineurs:
- Mise en évidence d’un marqueur clonal ou absence d’évidence pour une thrombocytose réactionnelle.