Leucémie myéloïde chronique (LMC)
Brève description:
La leucémie myéloïde chronique est habituellement une maladie de l'adulte (médiane d'âge: 50 ans). Elle fait partie des syndromes myéloprolifératifs. L'évolution comprend une phase chronique de durée variable suivie d'une phase d'accélération et de la crise blastique terminale. Comme pour la leucémie lymphoïde chronique, le diagnostic est très souvent posé de manière fortuite à la faveur d'un examen sanguin. Dans 90-95% des cas, la cytogénétique révèle une translocation (9;22), appelée chromosome Philadelphie (Ph). Le gène chimérique BCR/ABL est toujours présent. Il est mis en évidence par biologie moléculaire. La leucémie myéloïde chronique atypique (Ph-, BCR/ABL-) fait partie des syndromes myélodysplasiques / myéloprolifératifs selon la classification OMS.
Aspect clinique:
Les formes symptomatiques de la leucémie myéloïde chronique sont caractérisées par de l'asthénie, des sudations nocturnes, une perte de poids ainsi qu'une sensation de gêne abdominale en rapport avec la splénomégalie. Une tendance aux ecchymoses spontanées est possible. La durée de la phase chronique est en moyenne de 3.5 ans; elle peut s'échelonner entre 1 à 10 ans. En règle générale, la phase d'accélération dure quelques mois à une année. La crise blastique est identique à une leucémie aiguë de novo dans sa présentation avec cependant la particularité d'être résistante aux traitements. Un tiers des acutisations est de type lymphoblastique.
Traitement:
Jusqu’en 2000, le traitement de la LMC (leucémie myéloïde chronique) était basé sur l’hydroxyurée et l’alpha-interféron avec des taux de réponse insatisfaisants chez la plupart des patients. Malgré le traitement, les patients développaient une phase accélérée après env. 5 ans en moyenne, puis une crise blastique seulement quelques mois plus tard. Celle-ci connaissait presque toujours une issue rapidement fatale.
L’arrivée de l’inhibiteur de la tyrosine kinase imatinib par voie orale (Glivec®) a révolutionné le traitement de la LMC. L’imatinib inhibe la BCR-ABL kinase active constitutive et bloque ainsi la prolifération incontrôlée des cellules de LMC. Il s’agit du premier exemple réussi de traitement ciblé ("targeted therapy") capable d’interrompre le mécanisme pathogénique d’une maladie avec un inhibiteur de petit poids moléculaire. Sous imatinib, 97% des patients parviennent à une réponse hématologique complète. La survie à 10 ans atteint 84%. Les décès dus à la LMC sont aujourd’hui devenus rares. Les nouveaux inhibiteurs de l’ABL, tels que le nilotionib (Tasigna), le dasatinib (Sprycel), le bosutinib (Bosulif) ou le ponatinib (Iclusig) sont encore plus puissants et sont parfois même efficaces dans les cas réfractaires à l’imatinib. La greffe de cellules souches allogène n’a plus une place aujourd’hui que chez les enfants et les patients réfractaires au traitement.
Aspect hématologique:
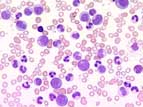
Dans la leucémie myéloïde chronique, des comptes leucocytaires supérieurs à 300 x 109/l sont possibles. A l'examen du frottis sanguin, tous les intermédiaires immatures de la granulopoïèse sont présents. Les éosinophiles et les basophiles sont habituellement augmentés. Une thrombocytose est fréquente. Une anémie apparaît lorsque les numérations leucocytaires sont très élevées. Le score de la phosphatase alcaline leucocytaire est effondré.
Aspect de la moelle osseuse:
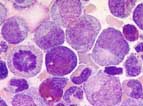
La moelle osseuse est riche; on note une prédominance de la granulopoïèse ainsi qu'une augmentation du nombre de mégacaryocytes. Fréquemment, la proportion d'éosinophiles et de basophiles est élevée. On peut observer parfois la présence de "sea-blue histiocytes" (histiocytes bleus); ces éléments ne sont pas pathognomoniques de la leucémie myéloïde chronique; on peut les rencontrer dans des pathologies aussi diverses que le purpura thrombopénique immun (PTI), la drépanocytose ou encore des maladies lysosomiales d'accumulation (Niemann-Pick, Tay-Sachs).
Consultez également la classification OMS des hémopathies malignes.
Diagnostic:
Le diagnostic d’une LMC peut être posé selon la définition de l’OMS sur la présence simultanée d’un gène de fusion BCR-ABL et d’une prolifération granulopoïétique mature dans le sang ou la moelle. Un gène de fusion BCR-ABL se trouve aussi, sur le plan du diagnostic différentiel, dans certains cas de leucémie aiguë lymphoblastique (LAL) de type B et occasionnellement dans la leucémie myéloïde aigu (LMA). Dans de rares cas, une LMC peut aussi se manifester par une thrombocytose isolée, raison pour laquelle il faut toujours exclure un BCR-ABL par une RT-PCR. En cas de suspicion de LMC, on fait une recherche de BCR-ABL par RT-PCR dans le sang. Un examen de la moelle avec cytogénétique fait obligatoirement partie du bilan initial. Du point de vue cytogénétique, on trouve dans 90-95% des cas une translocation balancée (9;22), autrement dit un chromosome de Philadelphie (Ph). Certaines autres altérations cytogénétiques fournissent des informations pronostiques importantes.