Essential thrombocythemia (ET)
Abstract:
Essential thrombocythemia belongs to the myeloproliferative neoplasms. It affects only the megakaryopoietic cell line. It is characterized by a persistent increase of the platelets > 450 x 109/L. Values can be increased beyond 1000 x 109/L. The function of the platelets is often impaired and can lead to bleeding and/or arterial and venous thrombosis. In about 60% of cases, a JAK2 V617F-Mutation, in 25% a mutation in the gene of calretikulin (CALR) and in 2% a mutation of the thrombopoietin receptor MPL can be demonstrated.
Clinical picture:
On average, the patients are more than 60-years-old at the time of diagnosis. The incidence is ca. 1.0/100'000/year. The patients may be asymptomatic and the diagnosis is in over 50% of cases established by chance. Symptoms usually result from platelet dysfunction in the microcirculation and manifest themselves in alternating neurologic complaints such as burning sensations of the soles of the foot (erythromelalgia), headaches and dizziness. Arteriolar thrombi lead to acrocyanosis and ischemic damage (e.g. cerebrovascular thrombosis, myocardial infarction). Venous thrombosis of liver veins can cause a Budd-Chiari-syndrome. In patients with increased risk for thromboembolic events, hydroxurea can reduce the frequency of such compliactions. It is important to differentiate ET from the many forms of secondary thrombocytosis. These have no association with thrombotic complications
Pathomechanism:
JAK2 V617F, MPL-mutation and CALR-mutations are so called "driver"-mutations that invoke directly the phenotype of increased megakaryopoiesis. The thrombopoietin receptor MPL and JAK2 are part of the signaling pathway that drives megakaryopoiesis. The exact mechanism of the CALR-mutation is unclear. It is supposed that in a part of patients additional mutations are necessary to provoke the disease. It is insufficiently understood why the same mutations evoke different clinical pictures (PMF, ET oder PV).
Therapy:
In order to prevent thrombotic complications the plarelet count must be reduced. Hydroxyurea (Litalir®), anagrelid (Xagrid®, Thromboreductin®) and interferon-alpha can be used therfor.
Hematology:
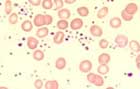
Platelets are markedly elevated (> 450 x 109/L to 1500 x 109/L). Moreover, mean platelet volume is increased with anisocytosis of the platelets and giant platelets with hypogranular and agranular forms. Occasionally, megakaryocyte remnants are seen. Basophilia is typical but not marked. Mild leukocytosis up to 20 x 109/L can occur. Higher values argue against ET.
Bone marrow:
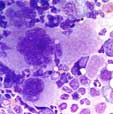
The megakaryopoiesis is increased with hyperlobulated nuclei (staghorn-like). In the biopsy, loose groups of megakaryocytes are seen but no dense clusters. The cells look rather monomorphic, small forms are not increased. Cellularitiy of the bone marrow is typically normal, but can be slightly increased in JAK2V617F-positive ET. A clearly increased cellularity argues against the diagnosis of ET and is more compatible with the hypercellular phase of primary myelofibrosis. A relevant fibrosis also argues against the diagnosis of ET.
Diagnosis:
According to the WHO 2016-classification, diagnosis of ET requires meeting all 4 major criteria or the first 3 major criteria and the minor criterion
Major criteria:
- Platelet count > 450 x 109/L
- BM biopsy showing proliferation mainly of the megakaryocyte lineage with increased numbers of enlarged, mature megakaryocytes with hyperlobulated nuclei. No significant increase or left shift in neutrophil granulopoiesis or erythropoiesis and very rarely minor (grade 1) increase in reticulin fibers
- Not meeting WHO criteria for BCR-ABL11 CML, PV, PMF, myelodysplastic syndromes, or other myeloid neoplasms
- Presence of JAK2, CALR, or MPL mutation
Minor criterion:
- Presence of a clonal marker or absence of evidence for reactive thrombocytosis