Primary Myelofibrosis (PMF)
Abstract:
Myelofibrosis is a myeloproliferative neoplasm and is characterized by the proliferation of megakaryocytes and the granulopoietic lineage. The megakaryocytes produce various cytokines that induce an increase of fibroblasts and bone marrow fibrosis. Increasing bone marrow failure results with pancytopenia and leukoerythroblastic blood picture. The latter is a sign of extramedullary hematopoiesis, mainly in the spleen. Splenomegaly can be massive. In about 50% of patients, a JAK2 V617F-mutation, in 35% a mutation of calreticulin (CALR) and in 5-10% a mutation of the thrombopoietin receptor MPL is present.
Clinical picture:
Mean age of patients is 67 years. Only 5% of patients are younger than 40 and 17% younger than 50 years. The incidence is about 0.5/100'000/year. 30% of patients are asymptomatic at the time of diagnosis, e.g. during a check-up, splenomegaly is found leading to the diagnosis. A splenomegaly is present in > 95% of patients. Often, the spleen is huge reaching down to the pelvis. Patients suffer from bloating, abdominal pain and an early feeling of being sated. Hepatomegaly is present in 40% of cases. Characteristic for PMF are pronounced general complaints such as fatigue, weakness, shortness of breath, weight loss, night sweats, fever and pruritus. As in essential thrombocythemia, arterial and venous thrombosis occurs. About 4% of cases develop acute myeloid leukemia.
Pathomechanism:
JAK2 V617F-mutation, MPL-mutation and CALR-mutation are driver-mutations, that evoke directly the phenotype of increased megakaryocytopoiesis. The thrombopoietin receptor MPL and JAK2 are part of the signaling pathway that drives the megakaryocytopoiesis. The exact mechanism of the CALR-muatation is unclear. It is insufficiently understood why the same mutations lead to different diseases (PMF, ET or PV).
Therapy:
The only curative therapy of PMF is allogenic bone marrow transplantation that is only considered in some of the patients with advanced disease. Splenomegaly and the general complaints can be successfully treated with the JAK1/2-inhibitor ruxolitinib (Jakavi@). Thus, quality of life of patients can be much improved .
Hematology:
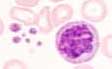
In the early phase, there is a thrombocytosis and neutrocytosis, in the later course pancytopenia. Typically, there is a leukoerythroblastic blood picture with normoblasts, marked anisocytosis, and poikilocytosis of erythrocytes with tear drop cells. Occasionally, megakaryocyte remnants are found. The thrombocytes are hypogranular or agranular and enlarged with giant platelets. In the late phase, myeloid precursor cells and blasts are increasingly present.
Bone marrow:
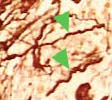
Due to fibrosis, a bone marrow aspiration yields no material ("dry tap"), therefore, a bone marrow biopsy is necessary. In the early phase, the biopsy demonstrates increased megakaryocytopoiesis and granulopoiesis. With the progression of the disease, increasing hypocellularity and progressive fibrosis are observed. In the end phase, hematopoietic cells can be completely lacking.
Megakaryocytes are markedly increased and sit closely in large clusters. Striking is their vicinity to vessels and trabecula. Morphologically, the megakaryocytes are large and have hyperlobated , often with hyperchromatic nuclei due to chromatin agglutination. The nucleus/cytoplasm-ratio is increased. Typical is the "cloud-like" aspect. Dilated bone marrow sinus with intravascular hematopoietic cells are characteristic. Bone trabecula can be thickened.
Diagnosis:
According to the WHO 2016-classification, the diagnosis of overt PMF requires meeting all 3 major criteria, and at least 1 minor criterion.
Major criteria:
- Presence of megakaryocytic proliferation and atypia, accompanied by either reticulin and/or collagen fibrosis grades 2 or 3
- Not meeting WHO criteria for ET, PV, BCR-ABL1 CML, myelodysplastic syndromes, or other myeloid neoplasms
- Presence of JAK2, CALR, or MPL mutation or in the absence of these mutations, presence of another clonal marker,† or absence of reactive myelofibrosis‡
Minor criteria:
- Anemia not attributed to a comorbid condition
- Leukocytosis > 11 x 109/L
- Palpable splenomegaly
- LDH increased to above upper normal limit of institutional reference range
- Leukoerythroblastosis