Polycythemia vera (PV)
Abstract:
Polycythemia vera is a myeloproliferative syndrome with a predominant feature of increased erythropoiesis (increase of the hemoglobin concentration and hematocrit). In almost all cases, a somatic activating mutation of the janus kinase 2 (JAK2V617F) or a functionally equivalent mutation is present. This leads to hyperproliferation not only of erythropoiesis but also of granulopoiesis and megakaryocytopoiesis. Besides hyperviscosity, arterial and venous thrombosis is feared. PV usually occurs after the age of 60, however, it can also occur in younger patients. The incidence is about 0.8/100'000/year.
Clinical picture:
The increased hematocrit causes hyperviscosity of the blood. Typical symptoms include headaches, dizziness, lethargy, sweating, pruritus (particularly after showering = aquagenic pruritus) and redness of the head (plethora). The increased risk for arterial and venous thrombosis is important. Especially feared are myocardial infarctions, ischemic cerebrovascular infarctions and venous thromboses of the portal vein, the mesenteric veins and the splenic vein. If an additional defect of platelet function is present, a tendency toward bleeding could also be present. Hyperuricemia occurs frequently due to the increased cell turnover and can lead to attacks of gout. Splenomegaly is present in 75% of the patients. About 30% of patients develop a myelofibrosis (post-PV-MF). Unlike chronic myeloid leukemia, a transition to acute leukemia is less common.
Pathomechanism:
The role of JAK2 as signaling molecule in several receptor systems such as the erythropoietin receptor, the thrombopoietin receptor (MPL) and the G-CSF-receptor explains the increase of all myeloid cell line by the activating mutation V617F and thus the phenotyp of PV. It is unclear if this mutation alone is sufficient to trigger the disease. It is assumed that at least a part of the patients have additional mutations prior to the JAK2-mutation and that theses contribute to the development of the disease.
Therapy:
The primary therapy consist in repeated phlebotomies. Thus, an iron deficiency is induced that inhibits erythropoiesis. Other therapeutic options are cytoreduction by hydroxyurea (Litalir®), as well as interferon-alpha. In therapy-refractory cases, the JAK1/2-inhibior ruxolitinib (Jakavi®) is used.
Hematology:
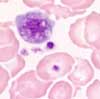
In addition to the increase of hemoglobin and hematocrit values (= polycythemia), an increase in the number of leukocytes and platelets is also found in most cases. Basophilia and/or mild eosinophilia is also typical. Myeloid precursor cells are rarely seen, blasts never occur. Since the therapy for polycythemia vera consists of repeated phlebotomy, microcytosis and hypochromia result as a sign of the desired iron deficiency.
Bone marrow:
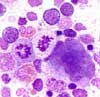
The bone marrow is hypercellular with predominance of erythropoiesis. Granulopoiesis and megakaryocytopoiesis are also increased ( "panmyelosis"). An increase in the basophils and eosinophils is also noticeable. Morphologically, the erythropoiesis and granulopoiesis are unobtrusive. The megakaryocytes are often found in loose groups close to trabecula and show a striking variance of size with small and large hyperlobated cells. In 20% of cases, there is mild reticulin fibrosis.
Diagnosis:
According to WHO 2016-classification, polycythemia vera can be diagnosed if all 3 major criteria or the first 2 major criteria and the minor criterion are present.
Major criteria
- Hemoglobin >165 g/L or hematocrit >49% in men, hemoglobin >160 g/L or hematocrit >48% in women or increased red cell mass (RCM)
- BM biopsy showing hypercellularity for age with trilineage growth (panmyelosis) including prominent erythroid, granulocytic, and megakaryocytic proliferation with pleomorphic, mature megakaryocytes (differences in size)
- Presence of JAK2V617F or JAK2 exon 12 mutation
Minor criterion
- Subnormal serum erythropoietin level