Immune thrombocytopenic purpura (ITP)
Abstract:
Thrombocytopenia is caused by anti-platelet autoantibodies or immune complexes. One distinguishes an acute and a chronic form as well as idiopathic and secondary (caused by another disease, e.g. CLL or systemic lupus erythematodes) forms.
Clinical picture:
The acute form usually occurs in children following a viral infection. Heavy bleeding is rare. A therapy is rarely needed. In over 90% of the children the thrombocytopenia disappears within weeks.
In adults, onset is either abrupt or slowly progressing. There is a peak incidence in young women and elderly people. The ratio of women and men is 3: 1. Even if the platelets fall under 5 x 109/L, fatal bleeding is rare. Symptoms are usually limited to generalized petechiae and easy bruising. Chronic ITP occurs idiopathically or secondarily in the case of lymphoproliferative syndromes and connective tissue diseases.
Therapy:
Therapy consists primarily in oral or intravenous corticosteroids. If heavy bleeding occurs intravenous immunoglobulins induce often a rapid increase of platelets. The response lasts often only for some weeks. In relapses or therapy resistant cases, splenectomy or immunotherapy with the anti-CD20-antibody Rituximab are options. Chronic courses can be treated with thrombopoietin-receptor-agonists such as Eltrombopag or Romiplostim.
Hematology:

In the blood film, platelets of variable size and thrombocytopenia are often distinct. Occasionally, giant platelets are found. Granulation of platelets is normal.
Bone marrow:
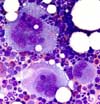
Markedly increased megakaryocytopoiesis is found in the bone marrow. Sometimes, the megakaryocytopoiesis is insufficiently increased in relation to the thrombocytopenia. Besides many young megakaryocytes with basophilic cytoplasm and monolobulated nucleus, the majority of megakaryocytes is large and mature with polylobulated nuclei.