Myélofibrose avec métaplasie myéloïde (MMM)
Brève description:
La myélofibrose avec métaplasie myéloïde fait partie des syndromes myéloprolifératifs. Elle est caractérisée par une fibrose de la moelle osseuse avec insuffisance de production compensée, du moins partiellement, par une hématopoïèse extramédullaire. L'anomalie clonale est localisée dans les mégacaryocytes qui sont augmentés et qui produisent des cytokines et des facteurs de croissance en excès (par exemple du PDGF = Platelet Derived Growth Factor, du facteur 4 plaquettaire) favorisant la prolifération des fibroblastes.
Aspect clinique:
Les patients ont généralement plus de 50 ans. L'affection débute de manière insidieuse par de la fatigue et une perte de poids. La splénomégalie est typique; une rate volumineuse peut être la cause de douleurs abdominales. Une hépatomégalie est également fréquente. Sur des clichés radiologiques du squelette, on peut observer un épaississement de l'os cortical.
Mécanisme pathogénique:
La mutation JAK2 V617F, MPL et les mutations CALR sont des mutations dites «driver», qui induisent directement le phénotype d’une mégacaryopoïèse augmentée. Les récepteurs de la thrombopoïétine MPL et JAK2 font partie de la principale voie de signalisation activatrice de la mégacaryopoïèse. Le mécanisme précis des mutations CALR n’est pas encore connu. On admet aujourd’hui qu’il faut chez au moins une partie des patients d’autres mutations pour déclencher la maladie. Ce qui n’est pas encore bien compris, c’est pour quelle raison des mutations identiques peuvent donner lieu à des maladies différentes (MFP, TE ou PV).
Traitement:
Le seul traitement curatif est la greffe de cellules souches allogènes, qui n’entre cependant en ligne de compte que dans les stades avancés chez certains patients. La splénomégalie et les symptômes généraux peuvent être traités efficacement avec l’inhibiteur JAK1/2 ruxolitinib (Jakavi). La qualité de vie des patients peut ainsi être considérablement améliorée.
Aspect hématologique:
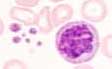
La myélofibrose avec métaplasie myéloïde est caractérisée par une anémie avec la présence de dacryocytes (cellules en larmes) et d'une érythroblastomyélémie. Des fragments nucléaires de mégacaryocytes sont souvent présents.
Aspect de la moelle osseuse:
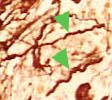
La ponction-aspiration de moelle ne ramène le plus souvent pas de matériel en raison de la fibrose. On parle de ponction blanche (punctio sicca). Durant la phase précoce, le matériel aspiré est marqué par une mégacaryopoïèse et une granulopoïèse augmentées.
Au fil de l’évolution de la maladie, la biopsie de moelle révèle d’abord une cellularité et une granulopoïèse augmentées, puis une hypocellularité progressive avec une fibrose de réticuline et de collagène croissante. Au stade terminal, les cellules hématopoïétiques peuvent être presque totalement absentes.
Les mégacaryocytes sont fortement augmentés et sont présents sous forme de grands amas denses. La proximité de ces amas avec les vaisseaux et les travées est également frappante. Du point de vue morphologique les mégacaryocytes sont de grande taille et hyperlobulés avec souvent des noyaux hyperchromatiques par agglutination de la chromatine. Le rapport noyau/cytoplasme est augmenté. Les cellules présentent typiquement un aspect nébuleux ou en ballon (mégacaryocytes «cloud like»). Les sinus médullaires dilatés contenant des cellules hématopoïétiques, en particulier des mégacaryocytes, sont également caractéristiques. Les travées osseuses peuvent être épaissies.
Diagnostic:
D’après la classification OMS, le diagnostic de myélofibrose primitive peut être retenu lorsque les trois critères majeurs et au moins un critère mineur sont vérifiés.
Critères diagnostics majeurs
- Hyperplasie mégacaryocytaire avec mégacaryocytes atypiques et fibrose de réticuline ou de collagène
- Exclusion d’une polycythémie vraie, d’une thrombocytémie essentielle, d’une leucémie myéloïde chronique BCR-ABL 1, d’un syndrome myélodysplasique ou d’une autre leucémie myéloïde
- Mise en évidence de JAK2 V617F, d’une mutation de MPL ou CALR ou d’un autre marqueur de clonalité; si négatif: Exclusion de causes secondaires
Critères diagnostics mineurs
- Formule sanguine leucoérythroblastique
- Leucocytose > 11 x 109/L
- Augmentation de la LDH
- Anémie qui ne peut pas être attribuée à une maladie concomitante
- Splénomégalie palpable