Polycythaemia vera (PV) / Polyglobulie primitive / Maladie de Vaquez
Brève description:
La polycythaemia vera fait partie des syndromes myéloprolifératifs. L'anomalie clonale touche essentiellement l'érythropoïèse; elle se traduit par une augmentation du taux d'hémoglobine et de l'hématocrite. Cette affection se voit surtout chez des individus de plus de 60 ans; elle n'épargne pas pour autant des sujets plus jeunes.
Aspect clinique:
Une augmentation de la viscosité sanguine est la conséquence d'un volume globulaire élevé. L'aspect du patient est pléthorique; la symptomatologie comprend classiquement des céphalées, des vertiges, des sudations et un prurit (en particulier après un bain ou une douche: prurit à l'eau). Les manifestations thrombo-emboliques sont fréquentes. En raison d'anomalies des fonctions plaquettaires, des complications hémorragiques sont possibles. L'hyperuricémie, due à un intense renouvellement cellulaire, est responsable de crises de goutte. Une splénomégalie est présente chez 75% des patients. En opposition avec la leucémie myéloïde chronique, la transformation en leucémie aiguë myéloïde est rare.
Mécanisme pathogénique:
Le rôle de JAK2 comme molécule de transmission de signaux dans plusieurs systèmes de récepteurs, tels que le récepteur de l’érythropoïétine, le récepteur de la thrombopoïétine (MPL) et le récepteur G-CSF, explique l’augmentation des trois lignées cellulaires myéloïdes sous l’effet de la mutation activatrice V617F et donc du phénotype de la polycythémie vraie. Nous ne savons par contre pas pour l’instant si la mutation suffit à elle seule à déclencher la maladie. On admet qu’il existe chez au moins une partie des patients des mutations supplémentaires qui précèdent la mutation JAK2 et contribuent à la survenue de la maladie.
Traitement:
Le traitement primaire consiste en phlébotomies répétées. La carence en fer qui en résulte inhibe l’érythropoïèse. La cytoréduction en général par hydroxyurée (Litalir) et l’interféron alpha sont d’autres options thérapeutiques. Dans les cas réfractaires, on utilise l’inhibiteur de JAK1/2 ruxolitinib (Jakavi).
Aspect hématologique:
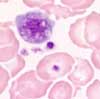
En-dehors de l'augmentation des valeurs de l'hémoglobine et de l'hématocrite, on constate souvent une leucocytose et/ou une thrombocytose. Une basophilie et/ou une discrète éosinophilie sont fréquentes. Après saignées répétées, une hypochromie est possible (carence en fer).
Aspect de la moelle osseuse:
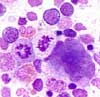
La moelle osseuse est caractérisée par une hyperplasie érythroïde. Les mégacaryocytes sont fréquemment augmentés; on constate parfois une élévation du nombre des éosinophiles et des basophiles.
Diagnostic:
D’après la classification OMS, on peut retenir le diagnostic de polycythémie vraie si les trois critères majeurs ou si les deux premiers critères majeurs plus le critère mineur sont vérifiés.
Critères diagnostics majeurs
- Hémoglobine >165 g/l ou hématocrite >49% (homme) / >160 g/l ou hématocrite >48% (femme) ou autre mise en évidence d’une augmentation du volume érythrocytaire (par ex. mesure du volume érythrocytaire par méthode de médecine nucléaire)
- Biopsie de moelle avec hypercellularité rapportée à l’âge et prolifération des trois lignées cellulaires (panmyélose) avec prolifération surtout érythrocytaire, granulocytaire et
- mégacaryocytaire et mégacaryocytes matures pléïomorphe (variation de taille)
- Mise en évidence d’une mutation JAK2 V617F ou d’une mutation fonctionnellement équivalente (par ex. mutation de l’exon 12 de JAK2)
Critères diagnostics mineurs
- Érythropoïétine sérique supprimée (nettement au-dessous de la limite inférieure de la norme).